 The University of Illinois at Chicago (UIC) College of Medicine states that delays in the transport and delivery of nutrients, proteins, and signaling molecules within nerve cells may bolster the development of amyotrophic lateral sclerosis (ALS). In the study, researchers reportedly assessed how the mutated form of the protein SOD1 caused slowdown in cellular transport.
The University of Illinois at Chicago (UIC) College of Medicine states that delays in the transport and delivery of nutrients, proteins, and signaling molecules within nerve cells may bolster the development of amyotrophic lateral sclerosis (ALS). In the study, researchers reportedly assessed how the mutated form of the protein SOD1 caused slowdown in cellular transport.
The results indicate that the mutated protein activated molecules called p38 kinases, which then modified a major motor protein linked to moving cargo along the nerve axons. Researchers say the modified motor proteins moved poorly when compared to controls that were exposed to SOD1 proteins that were not mutated.
The transport in genetically altered mice was also exhibited by SOD1 through the same mechanism, according to a recent news release.
Scott Brady, PhD, UIC professor, head of anatomy and cell biology, study co-principal investigator, notes, “The pathways between SOD1 and the p38 kinases could provide interesting targets for therapeutic intervention in treating ALS, both for some genetic forms and the spontaneous forms, where malfunctioning SOD1 is also a contributing factor.”
Brady adds that as cell death is the final stage in ALS, “We wanted to understand the pathological process in neurons leading up to cell death.”
The release reports that the study results reinforce research previously conducted by Brady and his colleagues. During prior research, the team reportedly used high-resolution video microscopy of squid axons to show that a mutant variant of the protein significantly slowed down the transport of material from one end of the cell to the other.
The current study appears in the online journal PLOS ONE.
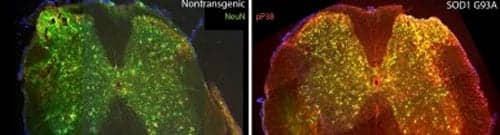
In photo above: Left- Mouse spinal cord with the normal form of SOD1 (neurons are labeled in green) Right- Mouse spinal cord with the mutated form of SOD1 (neurons where p38 kinase is activated are labeled in yellow)
Photo Credit: Rodolfo Gatto and Gerardo Morfini
Source: University of Illinois Chicago